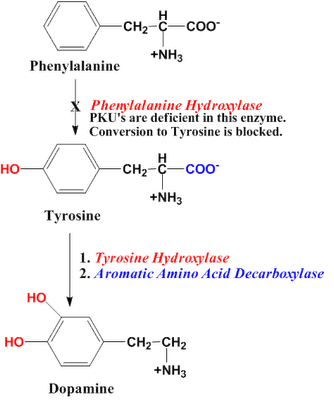
Autosomal recessive condition, where there is a deficiency in phenylalanine hydroxylase (breaks down phenylalanine into tyrosine), leading to accumulation of phenylalanine
Patients appear normal at birth, but manifest characteristic features during infancy
History / PE:
Urine has musty odor (from phenylacetic acid)
Fair skin, blue eyes, eczema
Diagnosis:
Elevated phenylalanine blood levels ( > 20mg/dL)
Guthrie test (qualitative, coloration test)
Normal tetrahydrobiopterin (confirm diagnosis)
Treatment:
Low phenylalanine diet
Avoid high protein foods
Complications:
Mental retardation
Seizures
Notes:
Guthrie test detects the metabolic products of phenylalanine in the urine